Neurofibromatosis type 2 is an autosomal dominant syndrome leading to the development of tumors in the neural system area. This disease is caused by mutations of the NF-2 gene which is the tumor suppresor gene located on chromosome 22 in the region q12. NF-2 gene encodes the schwannomin protein which functions as a negative growth regulator. So far, there have been described several dozen NF-2 gene mutations located in exons 1 -15 and in respective exon - intron joints.
Each type of the mutation was associated with its phenotypic manifestation. Missence mutations (mutations causing substitution of one or more amino acids) are mostly associated with a mild disease course, whilst mutations causing a frameshift or premature terminating of the reading frame correlate with a more serious course of the disease.
Examination
In our laboratory, we perform the mutation analysis of all fifteen exons of the NF-2 gene. We use PCR and direct sequencing using the capillary sequencer ABI PRISM 310.
-
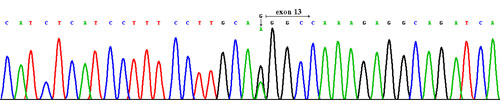 Fig.1
Fig.1Mutations of the NF2 gene. Heterozygous splice - site mutation c1341-1G>A in intron betveen the exons 12 and 13.
Clinical sensitivity:
The cause of the disease neurofibromatosis type 2 is the alterations in the gene NF2.
Sequence analysis of the coding exons and exon-intron connections of gene NF2 detects about 85% of the mutations listed in the publicly accessible version of the mutation database HGMD (The Human Gene Mutation Database).
MLPA (Multiple-Ligation Probe Amplification) analyzes mutations of a large-scale (amplification / deletion) it dstects about 14% of mutations in the NF2 gene listed in a publicly available version of the mutation database HGMD.
Analytical sensitivity and specificity of the sequencing: 99%.
Limitation:
Mutations deep in the introns and regulatory sequences are not detected. Rare polymorphisms at the site of priming or probes may cause a diagnostic error. In the case of mosaicism NF2 gene mutations will not be detected, if the altered cell line is not represented by at least 20%.
In the case of analysis of somatic mutations sequencing mutations will not be detected, if the altered cell line is not represented by at least 20%.